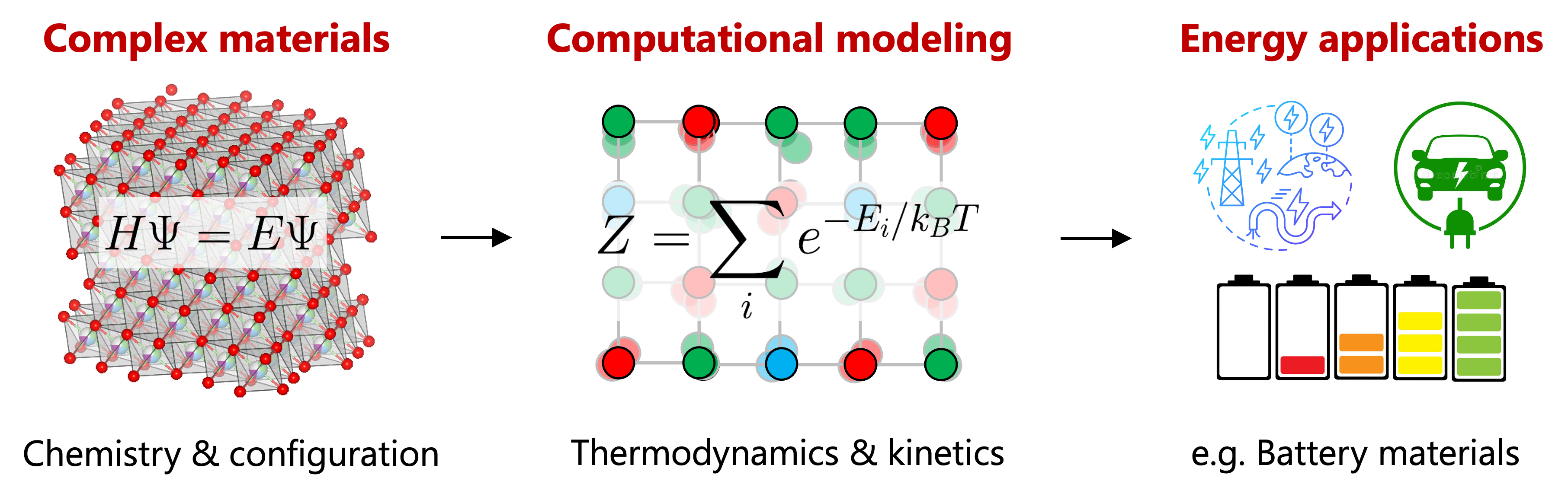
Materials modeling with atomistic simulations offers a powerful alternative to conventional trial-and-error methods by providing atomic-level insights for mechanistic understanding and accelerated materials discovery. A central objective is to predict material properties at finite temperatures, which requires computationally efficient statistical-mechanics approaches that retain ab initio accuracy.
Our research focuses on developing computational frameworks that connect first-principles descriptions of crystalline and molecular microstates with their macroscopic thermodynamic and kinetic properties.
By bridging length scales, time scales, and physical phenomena, these frameworks accelerate materials discovery while deepening our fundamental understanding of materials chemistry for technological development.
Currently, we are particularly interested in:
- Computational modeling of complex materials under realistic experimental conditions.
- Atomistic simulations that integrate statistical mechanics with first-principles calculations.
- Method development in AI for science: simulations for large and open systems, generative dynamics, and experimental alignment.